


Artificial Intelligence for facilitated drug discovery
The global pharmaceutical industry faces increasing business risks in connection with rising research costs, patent expiration, pricing issues, and constantly increasing competition, which, accordingly, reduces margins. The main focus area of market-driven healthcare companies is to allocate resources more wisely to reduce business risks and increase efficiency, competitiveness, and profitability.
Artificial intelligence and its subsets of Machine Learning, Deep Learning, as well as Artificial Neural Networks are being actively applied to optimize internal operations associated with drug discovery and manufacturing. The latest, cutting-edge technologies are used for faster, more efficient and significantly more cost-effective drug discovery and repurposing, as well as enhanced clinical trials.
The problem
A few decades ago, design, validation, as well as further synthetic procedures were manual and inefficient. Advances in computational technology combined with multi-omics data facilitated the active implementation of various bioinformatics solutions, but challenges still remain.
Most issues arising during drug discovery and manufacturing commonly include adverse absorption, distribution, metabolism, excretion, and toxicity properties, which interferes with research and development. This increases the interest in the early-stage prediction of the key properties of candidates so that the chances of success in creating new medicines can be notably increased.
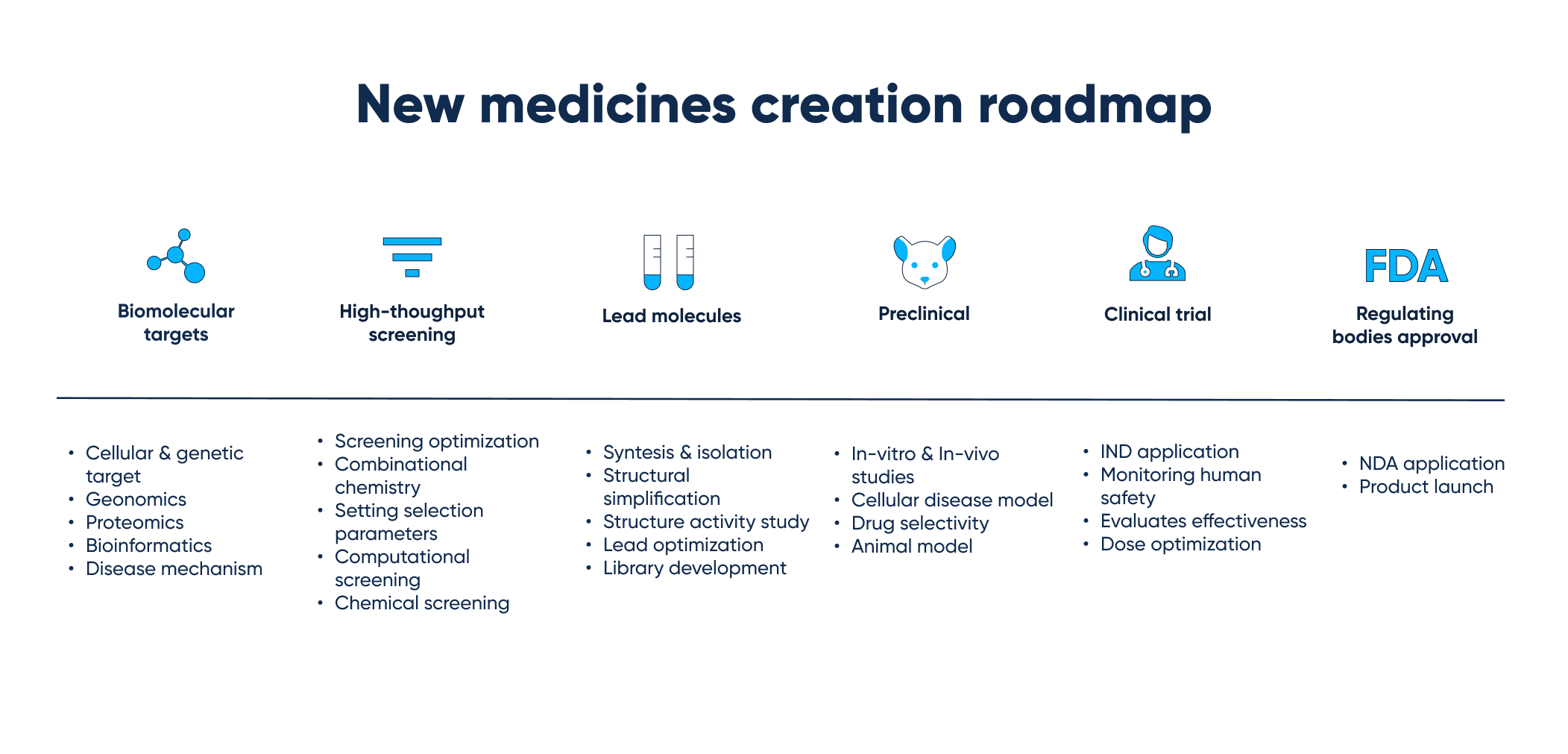
New medicines creation roadmap, from biomolecular targets up to regulating bodies’ approval and product launch
The solution
With the rapid advent of AI and its different subsets, the global pharmaceutical industry notably flourished. Biological and chemical data in the form of Big Data are being actively utilized for modern ML/DL approaches. These facilitate the identification of the undiscovered patterns and models that are further applied in studies. This way, ML/DL approaches are used for the simplified detection of therapeutically valuable compounds.
In 2019, the World Economic Forum has stated that implementing Artificial Intelligence together with Big Data will cause the fourth industrial revolution that could radically change the practice of modern scientific research. The global pharmaceutical market, like any other sector, is considering the many potential benefits of applying ML, DL, and ANN along with Big Data to overcome the most common issues associated with drug discovery.
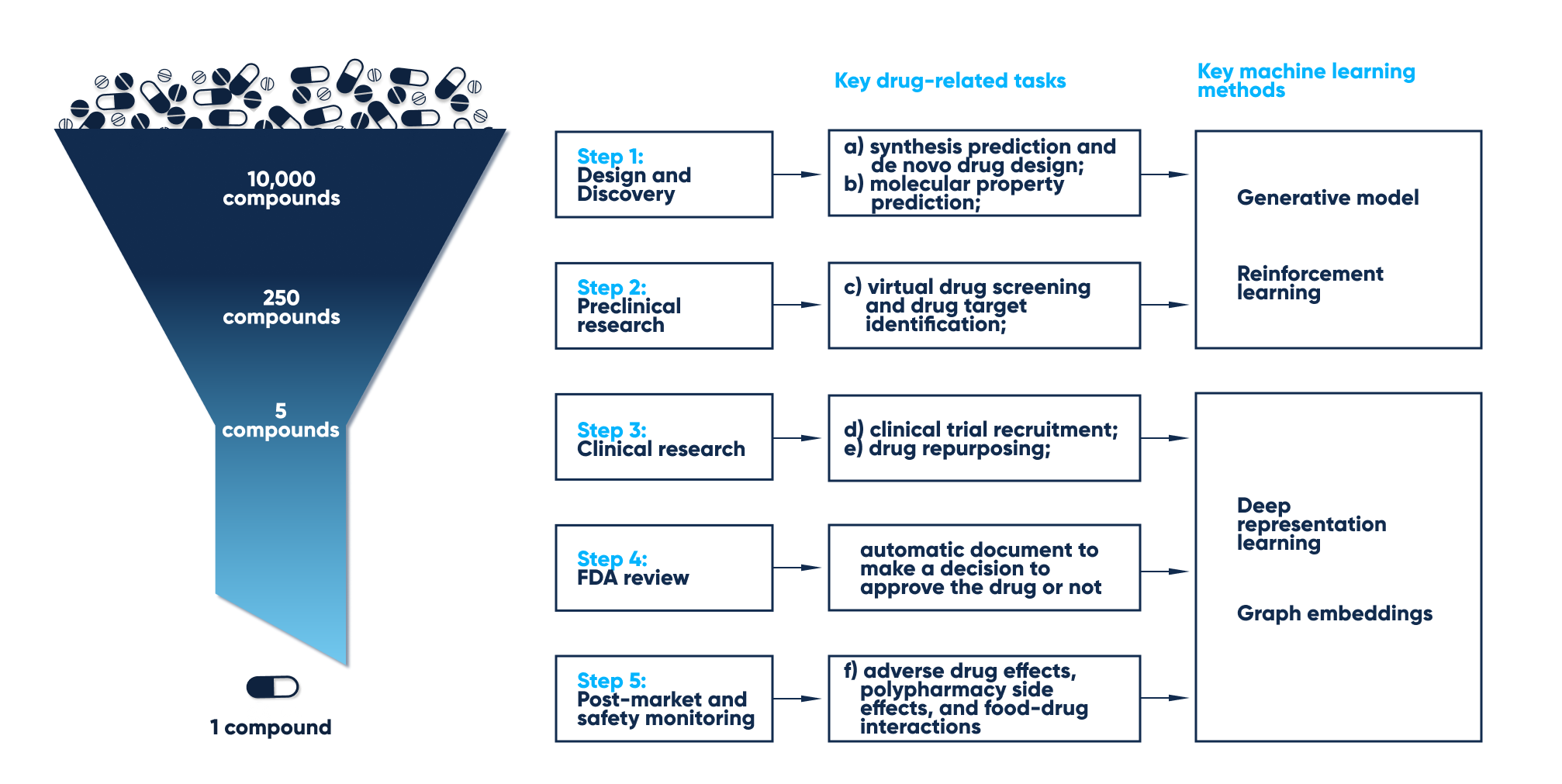
How Machine Learning helps prevent the most common issues associated with drug discovery
Computational drug discovery software for streamlined scientific research
Prediction of drug-target interactions
Drug-target interaction means interaction between various chemical compounds and bimolecular drug targets. This process is an important element of modern drug discovery, since the therapeutic effect of new pharmaceutical products is a direct result of the drug-target interaction.
Little and limited knowledge in DTI results from traditional experiments, conducted by laboratory assistants who utilize somewhat outdated investigation methods and equipment, which have been utilized for decades. Insufficient knowledge causes interest in finding new methods and designing modern equipment for more accurate prediction of DTI.
Traditional practices have numerous financial and technical limitations, which is why researchers switch to modern approaches:
- The ligand-based design approach (relies on understanding the molecules that bind to the biological target of interest)
- The chemo-genomic design approach (relies on combining sequence information with the protein structure to link to the chemical inhibitors)
- Text mining (the process of examining large collections of documents to discover new information)
- Modern methods taking advantage of ML and DL
Some methods of modern computational prediction utilizing ML and DL are described in the table below:
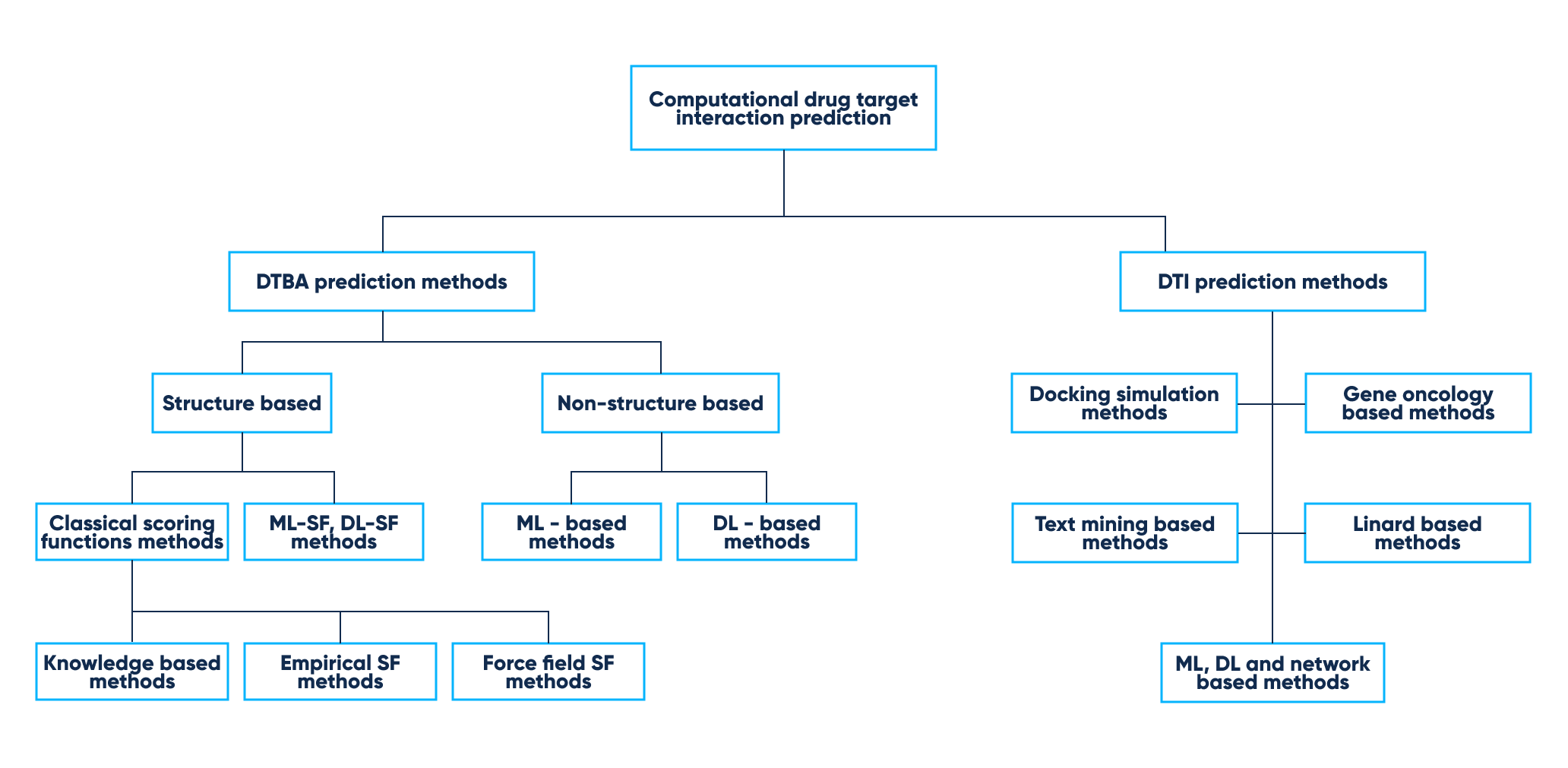
Computational drug target interaction prediction
Prediction of protein structures
Proteins are important biomolecules that have great influence on processes that happen in the human body. These include enzymatic activity, receptor activity, cell signaling, hormonal activity, and internal cell transport.
Protein structure explains the protein’s most important functions, its activity and its pathological conditions. Traditional methods of experimental data research shed light on about 100,000 unique protein sequences. However, that’s only a tiny part of the protein structures already known to mankind. To address this issue, researchers implement computational approaches that make the determination of unknown protein sequences a more accurate process.
One example is the AlphaFold network designed by DeepMind Technologies to determine protein structures. Detecting mutations that arose in response to other such mutations, it interprets spatial proximity to identify protein sequences.
To solve this issue, the system is implementing Deep Artificial Neural Networks, which help identify the evolutionary patterns of structural protein sequences associated with contact distribution and restraints. What’s more, with the help of Deep Learning, it calculates the potential of the protein structure.
Discovery of novel compounds
De novo molecule design is the complex process of creating molecular compounds to create new treatments. This process takes place with consideration of the drug-target binding affinity prediction, drug-target interaction and pharmacophore.
De novo molecule design has undergone major changes thanks to reinforcement learning, which is being used by researchers to generate new compounds with the desired physical, chemical, and biochemical properties. Reinforcement learning involves the analysis of possible effects on the human organism and evaluation of the possible results, which helps to determine the most efficient treatment.
Prediction of pharmacokinetic properties
Pharmacokinetic properties include absorption, distribution, metabolism, and excretion of a novel compound. The need for tools to precisely predict key pharmacokinetic properties is rising, as these provide researchers with a better understanding of how pharmaceutical compounds are interacting with the human organism. These should serve two important purposes. The right prediction tools might reduce exhaustion risks at the later stages, and optimize both screening and testing by predicting molecule profiles.
In today’s pharmaceutical research, Machine Learning is being actively utilized to predict ADMET properties. ADMET properties mean the absorption, distribution, metabolism, excretion, and toxicity of compounds. Accurate studies are possible due to the availability of an enormous amount of precise pharmacokinetic data. This helps to predict dose size, dosing frequency, blood-brain barriers, and other ADMET properties.
Software used in drug discovery process
AlphaFold
In 1972, Nobel laureate Christian Anfinsen came up with an interesting theory about amino acid sequences. This hypothesis has become the basis for research that continued for almost 50 years.
In 2018, DeepMind Technologies have introduced the first major version of the breakthrough system based on an Artificial Neural Network, which could accurately predict complex structures without using ready templates. By examining evolutionarily related biological sequences and utilizing the alignment of amino acid sequences together with pairwise representations, the system can refine the protein’s spatial graph.
For example, the system can refine the hemoglobin protein structure and its corresponding graph:
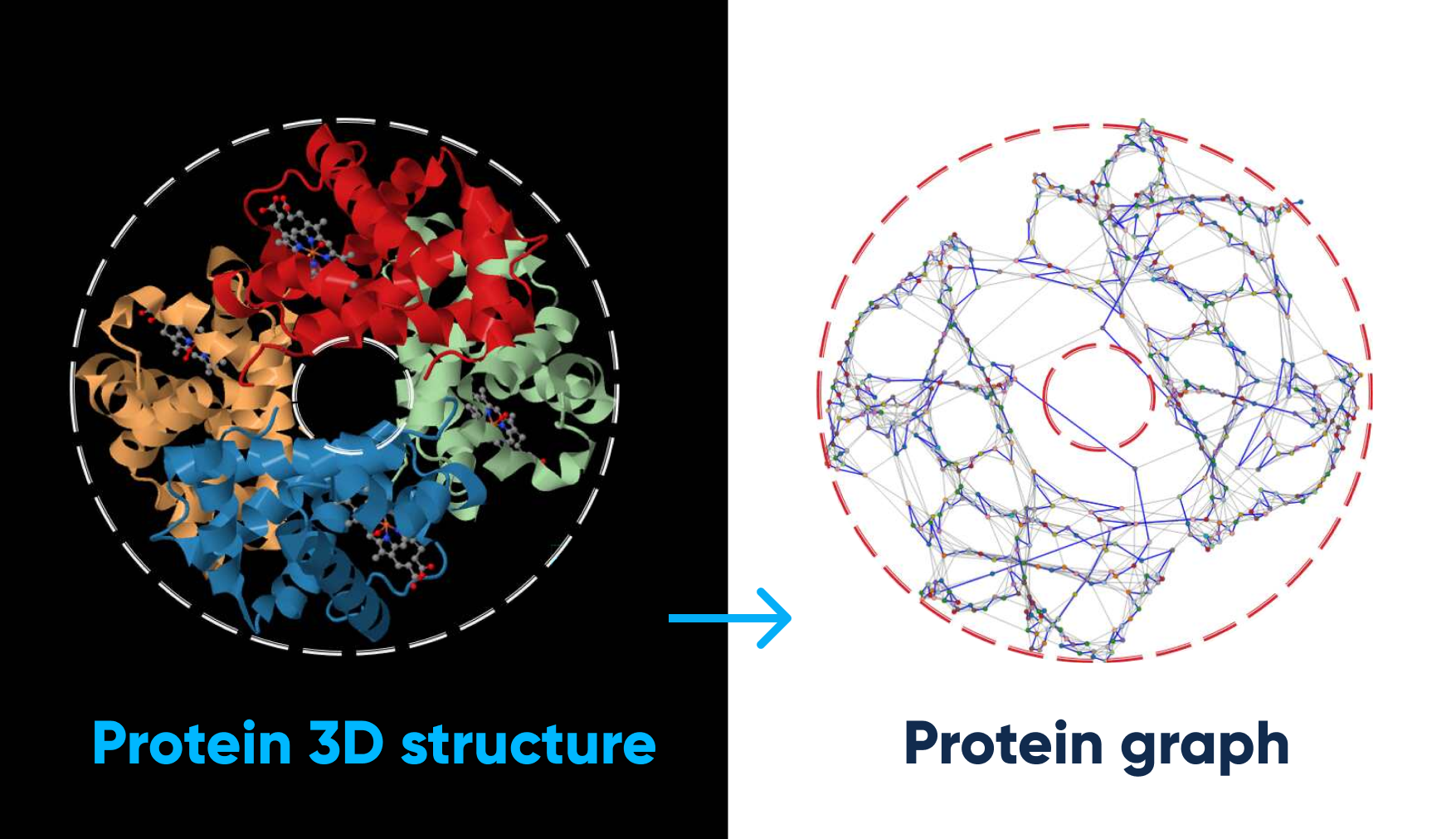
How system refines the hemoglobin protein structure and its corresponding graph. Image source
The introduced AlphaFold network is potentially a very useful invention to the pharmaceutical community. Notably expanding the number of known protein structures, the advanced AlphaFold system might provide new approaches for the efficient treatment of Alzheimer’s, Parkinson’s, ADHD, and other severe conditions.
Some researchers call the AlphaFold network one of the greatest scientific achievements of the 21st century. Others believe there are still more important challenges to address, and it won’t revolutionize drug discovery. One thing’s for sure – AlphaFold enjoys great interest in the scientific community, and there is more to come. And it’s already being actively applied.
Here’s a great example of how the system can create 3D shapes from a string of amino acids:
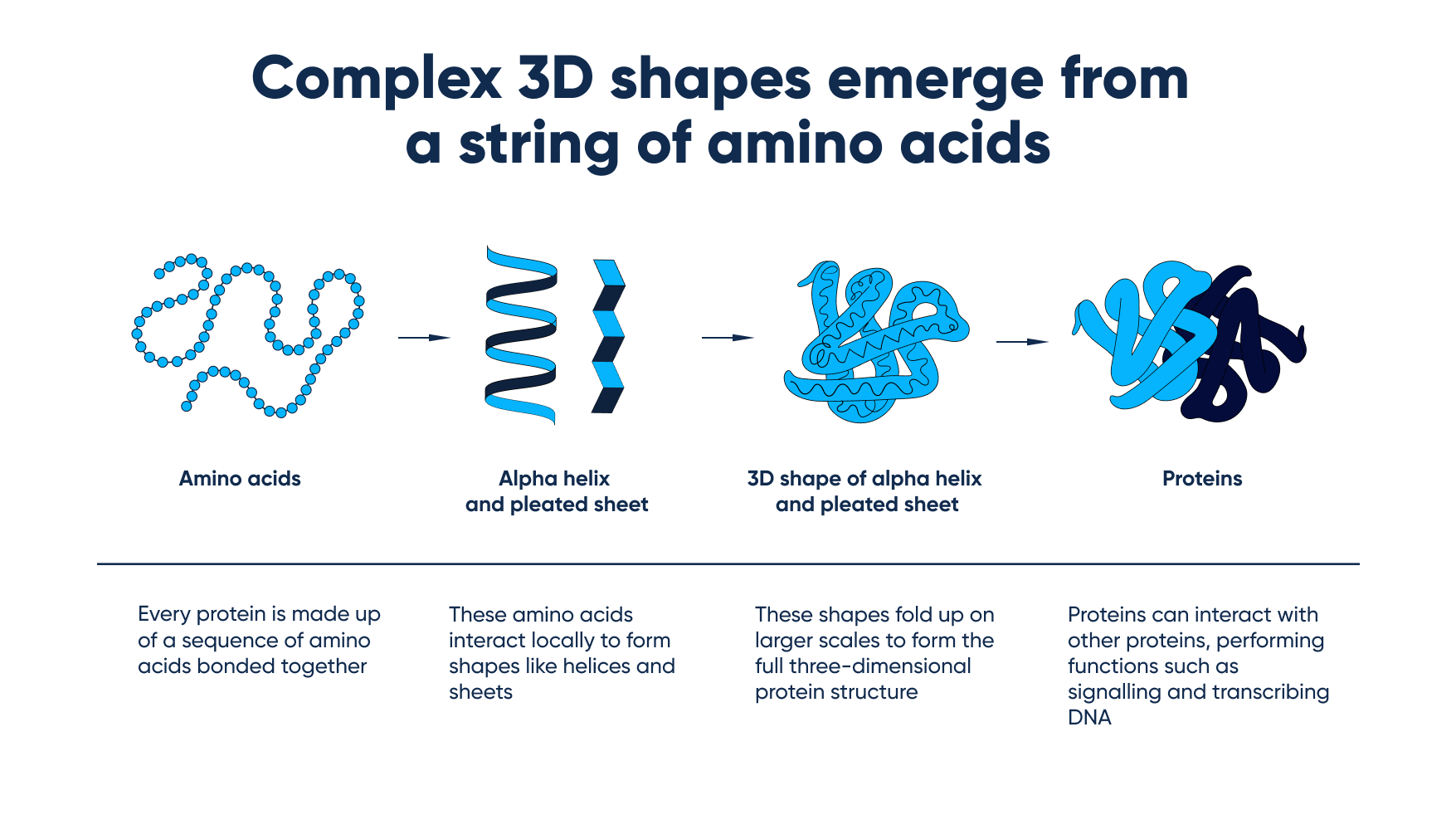
How the system creates 3D shapes from a string of amino acids
DeepChem
DeepChem is another solution worth mentioning when talking about facilitating drug discovery and design. DeepChem is an open-source, Python-based framework, which implements various frameworks such as Google’s TensorFlow, dmlc XGBoost, and scikit-learn, to popularize Deep Learning for accurate drug discovery, material science, quantum chemistry, and biology.
There are lots of detailed tutorials on using this tool suggested by:
Cyclica
Cyclica integrates two platforms (Ligand Design and Express) to accelerate the process of new drug discovery:
- Ligand Design performs exploration of the chemical space and implements selective methods to design new compounds;
- Ligand Express helps track target and non-target interactions between candidates (new compounds identified as potentially useful for further drug design), which helps to prioritize lead candidates.
Cyclica’s vision is to further expand its initiatives associated with Artificial Intelligence to drive more innovation. They’re focusing on shortening manufacturing timelines to make high-quality medication more accessible. Another thing, the organization is collaborating with over 100 global pharmaceutical and biotech companies. These specialize in multiple therapeutic areas including respiratory, oncology, ophthalmology, and other.
ODDT
The ODDT (Open Drug Discovery Toolkit) is an open-source toolkit for efficient, computer aided drug discovery. It implements state-of-the-art methods, which are Machine Learning scoring functions (RF-Score, NNScore), and provides interesting features, for instance, 3 popular molecular shape comparison methods (USR, USRCAT, and Electroshape).
The ODDT is a comprehensive toolkit, which can be used for cheminformatics, molecular modeling, and more. What’s more, it’s designed to be easily customizable, so that it can be extended by users.
Final words
Artificial Intelligence has long been proven to show notable progress in boosting pharmaceutical production. Modern advances help identify promising compounds and increase success rates.
We at Abto Software strongly believe there are numerous applications of innovative ML and DL approaches. And the main goal should be figuring out how these can boost your business.
We help pharmaceutical companies to facilitate:
- Drug development
- Drug simulation
- Laboratory inventory
- Sanitary control
- Clinical trials
- Incident reporting
By delivering:
- ERP (Enterprise Resource Planning) systems
- MRP (Material Requirements Planning) platforms
- Production planning and control software
- Product traceability and recall tools
And more!
By leveraging modern advances, pharma companies, pharmacy chains, and clinics can automate daily routines. This means decreased risks, increased overall employee satisfaction, thought-out allocated financial resources, and improved business profitability!
The key is to bring together software engineers who can get the most out of using computational technology. Contact us for custom software development!